library(mandala)
library(dplyr)
library(ggplot2)
library(Matrix)Two-Stage Genomic Analysis
Integrating GBLUP with two-stage MET workflows
Overview
This tutorial extends the two-stage MET workflow to include genomic information. By incorporating a genomic relationship matrix (GRM) at Stage 2, we can:
- Obtain genomic estimated breeding values (GEBVs)
- Predict performance of untested genotypes
- Leverage marker information for selection decisions
The key innovation is using GM(geno, Gmat) alongside vcov(RMAT) to combine genomic relationships with properly propagated BLUE uncertainty.
NoteLearning Objectives
- Prepare and align a GRM for Stage 2 analysis
- Fit GBLUP models using
GM()withvcov(RMAT) - Use the
mandala_gp()wrapper for streamlined genomic prediction - Extract and compare GEBVs from different approaches
- Understand when genomic prediction adds value
ImportantPrerequisites
This tutorial assumes completion of Tutorial 06: Two-Stage MET. You should understand:
- The two-stage workflow (Stage 1 BLUEs, Stage 2 analysis)
- How
vcov(RMAT)propagates BLUE uncertainty - Basic heritability and variance component concepts
Data Setup
We’ll use the same phenotypic data structure as Tutorial 06, plus a simulated GRM.
Simulate Phenotypic Data
set.seed(42)
n_env <- 4
n_geno <- 50
n_rep <- 2
n_row <- 10
n_col <- 10
# Create base data structure
df <- expand.grid(
env = paste0("E", 1:n_env),
geno = paste0("G", sprintf("%03d", 1:n_geno)),
rep = 1:n_rep
)
# Assign spatial positions
df <- df %>%
group_by(env, rep) %>%
mutate(
row = rep(1:n_row, length.out = n()),
col = rep(1:n_col, each = n_row)[1:n()]
) %>%
ungroup()
# Simulate effects
env_effects <- rnorm(n_env, mean = 0, sd = 5)
names(env_effects) <- paste0("E", 1:n_env)
geno_effects <- rnorm(n_geno, mean = 0, sd = 3)
names(geno_effects) <- paste0("G", sprintf("%03d", 1:n_geno))
# GxE interaction
gxe_effects <- matrix(rnorm(n_env * n_geno, mean = 0, sd = 2),
nrow = n_geno, ncol = n_env)
rownames(gxe_effects) <- names(geno_effects)
colnames(gxe_effects) <- names(env_effects)
# Build response
df <- df %>%
mutate(
yld = 100 +
env_effects[as.character(env)] +
geno_effects[as.character(geno)] +
sapply(1:n(), function(i) gxe_effects[as.character(geno[i]), as.character(env[i])]) +
rnorm(n(), mean = 0, sd = 2)
)
# Convert to factors
df$env <- as.factor(df$env)
df$geno <- as.factor(df$geno)
df$rep <- as.factor(df$rep)
df$row <- as.factor(df$row)
df$col <- as.factor(df$col)
df$block <- df$rep
head(df)# A tibble: 6 × 7
env geno rep row col yld block
<fct> <fct> <fct> <fct> <fct> <dbl> <fct>
1 E1 G001 1 1 1 111. 1
2 E2 G001 1 1 1 95.0 1
3 E3 G001 1 1 1 101. 1
4 E4 G001 1 1 1 102. 1
5 E1 G002 1 2 1 105. 1
6 E2 G002 1 2 1 96.3 1 Simulate Genomic Relationship Matrix
In practice, you would compute the GRM from marker data. Here we simulate one that reflects the true genetic effects.
# Simulate a GRM based on true genetic effects
# In practice, use markers and compute with A.mat() or similar
geno_names <- paste0("G", sprintf("%03d", 1:n_geno))
# Create a correlation structure based on genetic similarity
set.seed(123)
n_markers <- 1000
markers <- matrix(sample(0:2, n_geno * n_markers, replace = TRUE),
nrow = n_geno, ncol = n_markers)
rownames(markers) <- geno_names
# Compute realized relationship matrix (VanRaden method 1)
p <- colMeans(markers) / 2
P <- 2 * (p - 0.5)
Z <- scale(markers, center = TRUE, scale = FALSE)
GRM <- tcrossprod(Z) / (2 * sum(p * (1 - p)))
# Ensure positive definiteness
GRM <- GRM + diag(1e-6, nrow(GRM))
rownames(GRM) <- colnames(GRM) <- geno_names
# Preview
GRM[1:5, 1:5] G001 G002 G003 G004 G005
G001 1.310757677 0.007381282 -0.039952294 0.002639819 -0.07516880
G002 0.007381282 1.246970820 -0.022202203 -0.022161677 -0.03918231
G003 -0.039952294 -0.022202203 1.302247359 0.005476592 -0.03991177
G004 0.002639819 -0.022161677 0.005476592 1.320564804 -0.05810764
G005 -0.075168798 -0.039182312 -0.039911768 -0.058107638 1.33312765Stage 1: Per-Environment Analysis
This follows the same workflow as Tutorial 06.
# Stage 1 preparation
s1_prep <- stage1_prep(
df = df,
env_col = "env",
s1_fixed = yld ~ geno,
s1_random = ~ rep + row + col,
s1_classify_term = "geno",
response_var = "yld"
)
# Audit
stage1_audit(df, s1_prep) env rows nonmiss_response nonmiss_id n_id_levels
1 E1 100 100 100 50
2 E2 100 100 100 50
3 E3 100 100 100 50
4 E4 100 100 100 50# Run Stage 1
MET_stage1_run <- mandala_stage1(
df = df,
prep = s1_prep
)
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: (none)
- The ignored set: rep, row, col
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: (none)
- The ignored set: rep, row, col
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: (none)
- The ignored set: rep, row, col
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: (none)
- The ignored set: rep, row, col# Bundle outputs
bundle <- stage1_bundle(MET_stage1_run)
cat("Stage 1 complete. BLUEs extracted for", nrow(bundle$blues), "observations\n")Stage 1 complete. BLUEs extracted for 200 observationsStage 2: Prepare Data and Matrices
# Create Stage 2 data and RMAT
s2_inputs <- stage2_prep(
bundle = bundle,
df_name = "stage2_data",
vcov_name = "RMAT",
out_env = "env",
out_id = "geno",
out_resp = "yld"
)
s2_data <- s2_inputs$stage2_data
RMAT <- s2_inputs$RMAT
# Align row IDs
rownames(s2_data) <- paste0(s2_data$env, "|", s2_data$geno)
s2_data <- s2_data[rownames(RMAT), , drop = FALSE]
# Ensure Matrix format
if (!inherits(RMAT, "Matrix")) {
RMAT <- Matrix::forceSymmetric(Matrix::Matrix(RMAT, sparse = TRUE))
}
head(s2_data) env geno yld
E1|G001 E1 G001 109.6489
E1|G002 E1 G002 109.0377
E1|G003 E1 G003 113.4326
E1|G004 E1 G004 108.8727
E1|G005 E1 G005 107.4885
E1|G006 E1 G006 106.9830Prepare GRM for Stage 2
Use mandala_grm_prep() to align and scale the GRM for the Stage 2 genotype set.
# For this tutorial, we'll prepare the GRM manually
# In practice with a file: mandala_grm_prep(GRM = "path/to/grm.csv", ...)
# Subset GRM to genotypes in s2_data
genos_in_s2 <- unique(s2_data$geno)
GRM_subset <- GRM[as.character(genos_in_s2), as.character(genos_in_s2)]
# Add small ridge for numerical stability
lambda <- 1e-8
GRM_subset <- GRM_subset + diag(lambda, nrow(GRM_subset))
# Convert to Matrix class
GRM_subset <- Matrix::Matrix(GRM_subset, sparse = FALSE)
cat("GRM dimensions:", dim(GRM_subset), "\n")GRM dimensions: 50 50 cat("Genotypes in GRM:", nrow(GRM_subset), "\n")Genotypes in GRM: 50 Stage 2 with GRM (GBLUP)
Now we fit the Stage 2 model with genomic relationships using GM(geno, Gmat).
stage2_gblup <- mandala(
fixed = yld ~ env,
random = ~ GM(geno, Gmat),
R_formula = ~ vcov(RMAT),
data = s2_data,
matrix_list = list(RMAT = RMAT, Gmat = GRM_subset)
)Initial data rows: 200
Final data rows after NA handling: 200 --- EM Warmup Skipped. Starting AI-REML ---Starting AI-REML logLik = -554.8966
Iter LogLik Sigma2 DF wall Restrained
1 -544.5046 11.399 196 14:53:57 ( 0 restrained)
2 -537.1838 8.664 196 14:53:57 ( 0 restrained)
3 -529.6235 5.268 196 14:53:57 ( 0 restrained)
4 -529.3817 3.122 196 14:53:57 ( 0 restrained)
5 -529.3817 3.122 196 14:53:57 ( 0 restrained)
Converged at iter 5 by tolerance(s): delta_theta=0.00e+00, delta_loglik=0.00e+00
Main AI-REML Loop finished. Combining results.summary(stage2_gblup)Model statement:
mandala(fixed = yld ~ env, random = ~GM(geno, Gmat), data = s2_data,
matrix_list = list(RMAT = RMAT, Gmat = GRM_subset), R_formula = ~vcov(RMAT))
Variance Components:
component estimate std.error z.ratio bound %ch
GM(geno,Gmat) 8.099542 2.0611808 3.929564 P NA
R.sigma2 3.121872 0.9564478 3.264028 P NA
Fixed Effects (BLUEs) [first 5]:
effect estimate std.error z.ratio
(Intercept) 107.172182 0.2497989 429.03383
envE2 -10.622852 0.3532820 -30.06904
envE3 -5.485783 0.3532820 -15.52806
envE4 -4.448731 0.3532820 -12.59258
Converged: TRUE | Iterations: 5
Random Effects (BLUPs) [first 5]:
random level estimate std.error z.ratio
GM(geno,Gmat) G001 0.5412109 0.8429880 0.6420149
GM(geno,Gmat) G002 0.2130938 0.8409079 0.2534092
GM(geno,Gmat) G003 3.6903397 0.8425172 4.3801357
GM(geno,Gmat) G004 -1.6155665 0.8429473 -1.9165688
GM(geno,Gmat) G005 3.4369444 0.8432061 4.0760431
logLik: -529.382 AIC: 1062.763 BIC: 1069.320 logLik_Trunc: -349.270Extract GEBVs
gebvs <- mandala_predict(stage2_gblup, classify_term = "geno")
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: env, [num mean] head(gebvs) geno predicted_value std_error
1 G001 102.5741 0.8521956
2 G002 102.2459 0.8501380
3 G003 105.7232 0.8517299
4 G004 100.4173 0.8521554
5 G005 105.4698 0.8524114
6 G006 102.4981 0.8527647Using mandala_gp() Wrapper
For convenience, mandala_gp() wraps the GRM preparation and model fitting into a single call.
# Example using mandala_gp() with a GRM file
# In practice, you would provide a path to your GRM file
gp_fit <- mandala_gp(
GRM = GRM_subset, # Can also be a file path: "path/to/grm.csv"
data = s2_data,
gen_col = "geno",
fixed = yld ~ env,
random = ~ GM(geno, Gmat),
R_formula = ~ vcov(RMAT),
R_matrix = RMAT,
lambda = 1e-8,
scale = FALSE,
method = "sparse"
)
summary(gp_fit$fit)For this tutorial, we’ll demonstrate with the manual approach since we have the GRM in memory:
# The stage2_gblup model we already fit is equivalent
# Extract predictions
gebv_predictions <- mandala_predict(stage2_gblup, classify_term = "geno")
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: env, [num mean] head(gebv_predictions) geno predicted_value std_error
1 G001 102.5741 0.8521956
2 G002 102.2459 0.8501380
3 G003 105.7232 0.8517299
4 G004 100.4173 0.8521554
5 G005 105.4698 0.8524114
6 G006 102.4981 0.8527647Comparing Approaches
Let’s compare predictions from: 1. Phenotypic Stage 2 (no GRM) — Tutorial 06 approach 2. Genomic Stage 2 (with GRM) — GBLUP
Fit Non-Genomic Stage 2
stage2_pheno <- mandala(
fixed = yld ~ env,
random = ~ geno,
R_formula = ~ vcov(RMAT),
data = s2_data,
matrix_list = list(RMAT = RMAT)
)Initial data rows: 200
Final data rows after NA handling: 200 --- EM Warmup Skipped. Starting AI-REML ---Starting AI-REML logLik = -554.8966
Iter LogLik Sigma2 DF wall Restrained
1 -544.7874 11.399 196 14:53:58 ( 0 restrained)
2 -537.2573 8.664 196 14:53:58 ( 0 restrained)
3 -529.2477 5.217 196 14:53:58 ( 0 restrained)
4 -528.9685 3.034 196 14:53:58 ( 0 restrained)
5 -528.9685 3.034 196 14:53:58 ( 0 restrained)
Converged at iter 5 by tolerance(s): delta_theta=0.00e+00, delta_loglik=0.00e+00
Main AI-REML Loop finished. Combining results.pheno_preds <- mandala_predict(stage2_pheno, classify_term = "geno")
Notes:
- The predictions are obtained by averaging across the hypertable
calculated from model terms constructed solely from factors in
the averaging and classify sets.
- The simple averaging set: env, [num mean] Merge and Compare
# Phenotypic predictions
p_pheno <- pheno_preds[, c("geno", "predicted_value")]
names(p_pheno) <- c("geno", "pred_pheno")
# Genomic predictions
p_geno <- gebv_predictions[, c("geno", "predicted_value")]
names(p_geno) <- c("geno", "pred_genomic")
# Merge
comparison <- merge(p_pheno, p_geno, by = "geno")
# Correlation
cor_pg <- cor(comparison$pred_pheno, comparison$pred_genomic, use = "complete.obs")
cat("Correlation (phenotypic vs genomic):", round(cor_pg, 3), "\n")Correlation (phenotypic vs genomic): 1 ggplot(comparison, aes(x = pred_pheno, y = pred_genomic)) +
geom_point(alpha = 0.6, size = 2.5) +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
title = "Phenotypic vs Genomic Predictions",
subtitle = paste("r =", round(cor_pg, 3)),
x = "Predicted Value (Phenotypic)",
y = "Predicted Value (Genomic/GBLUP)"
) +
theme_minimal()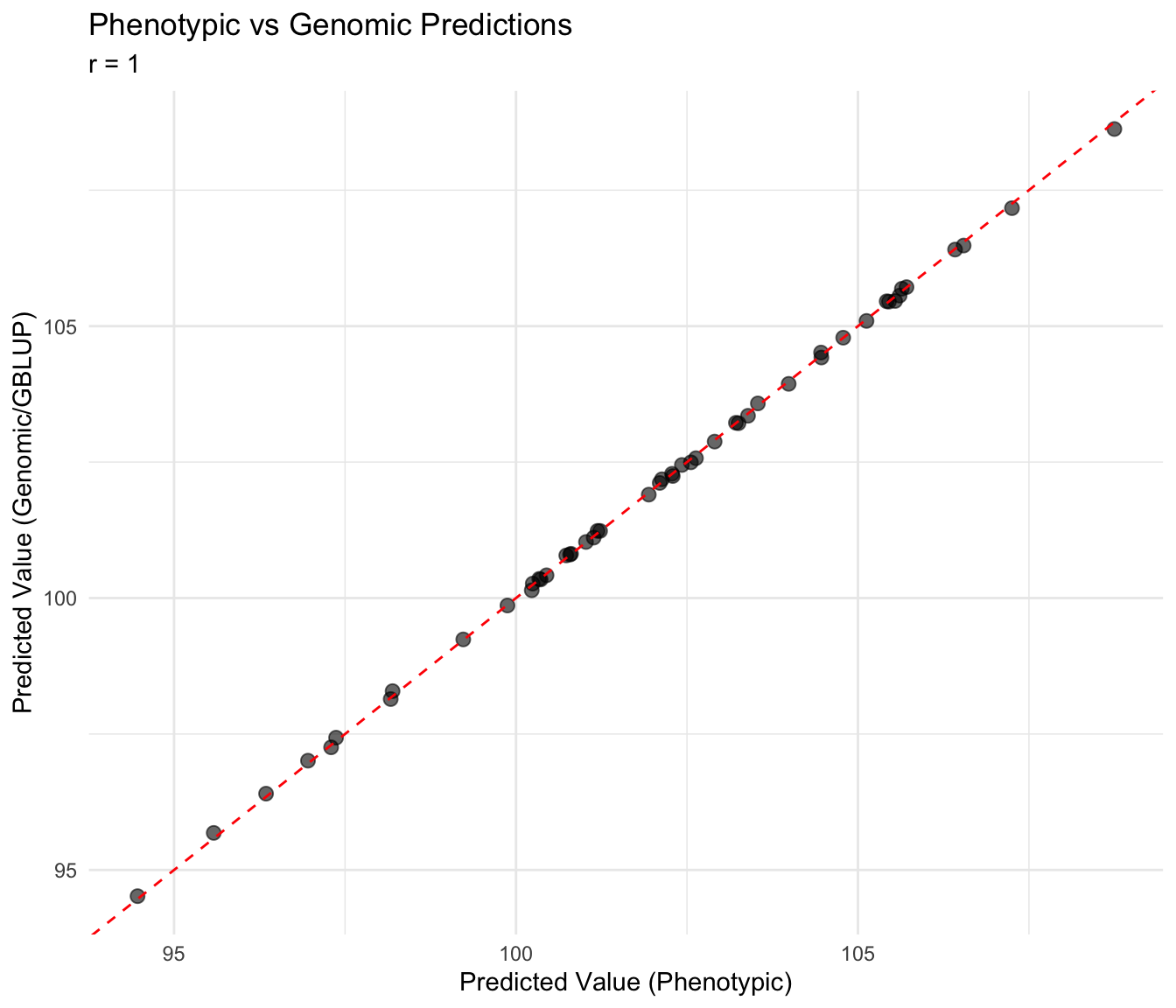
Variance Components Comparison
cat("=== Phenotypic Model Variance Components ===\n")=== Phenotypic Model Variance Components ===print(summary(stage2_pheno)$varcomp)Model statement:
mandala(fixed = yld ~ env, random = ~geno, data = s2_data, matrix_list = list(RMAT = RMAT),
R_formula = ~vcov(RMAT))
Variance Components:
component estimate std.error z.ratio bound %ch
geno 10.202405 2.6120170 3.905949 P NA
R.sigma2 3.033944 0.9498473 3.194139 P NA
Fixed Effects (BLUEs) [first 5]:
effect estimate std.error z.ratio
(Intercept) 107.191648 0.5145133 208.33601
envE2 -10.643971 0.3483644 -30.55413
envE3 -5.506118 0.3483644 -15.80563
envE4 -4.468908 0.3483644 -12.82826
Converged: TRUE | Iterations: 5
Random Effects (BLUPs) [first 5]:
random level estimate std.error z.ratio
geno G001 0.5955164 0.9465325 0.6291558
geno G002 0.2560207 0.9465325 0.2704828
geno G003 3.6784336 0.9465325 3.8862200
geno G004 -1.5921461 0.9465325 -1.6820828
geno G005 3.5115972 0.9465325 3.7099593
logLik: -528.969 AIC: 1061.937 BIC: 1068.493 logLik_Trunc: -348.857
component estimate std.error z.ratio bound %ch
1 geno 10.202405 2.6120170 3.905949 P NA
2 R.sigma2 3.033944 0.9498473 3.194139 P NAcat("\n=== Genomic Model Variance Components ===\n")
=== Genomic Model Variance Components ===print(summary(stage2_gblup)$varcomp)Model statement:
mandala(fixed = yld ~ env, random = ~GM(geno, Gmat), data = s2_data,
matrix_list = list(RMAT = RMAT, Gmat = GRM_subset), R_formula = ~vcov(RMAT))
Variance Components:
component estimate std.error z.ratio bound %ch
GM(geno,Gmat) 8.099542 2.0611808 3.929564 P NA
R.sigma2 3.121872 0.9564478 3.264028 P NA
Fixed Effects (BLUEs) [first 5]:
effect estimate std.error z.ratio
(Intercept) 107.172182 0.2497989 429.03383
envE2 -10.622852 0.3532820 -30.06904
envE3 -5.485783 0.3532820 -15.52806
envE4 -4.448731 0.3532820 -12.59258
Converged: TRUE | Iterations: 5
Random Effects (BLUPs) [first 5]:
random level estimate std.error z.ratio
GM(geno,Gmat) G001 0.5412109 0.8429880 0.6420149
GM(geno,Gmat) G002 0.2130938 0.8409079 0.2534092
GM(geno,Gmat) G003 3.6903397 0.8425172 4.3801357
GM(geno,Gmat) G004 -1.6155665 0.8429473 -1.9165688
GM(geno,Gmat) G005 3.4369444 0.8432061 4.0760431
logLik: -529.382 AIC: 1062.763 BIC: 1069.320 logLik_Trunc: -349.270
component estimate std.error z.ratio bound %ch
1 GM(geno,Gmat) 8.099542 2.0611808 3.929564 P NA
2 R.sigma2 3.121872 0.9564478 3.264028 P NAWhen Does Genomic Prediction Help?
Genomic prediction provides the most benefit when:
| Scenario | Benefit |
|---|---|
| Predicting untested genotypes | GRM allows prediction for genotypes without phenotypic data |
| Low heritability traits | Genomic info adds signal when phenotypes are noisy |
| Small family sizes | GRM captures relationships beyond pedigree |
| Selection accuracy | GEBVs often outperform phenotypic BLUPs |
TipCross-Validation
To assess prediction accuracy, use mandala_gp_cv() for cross-validation:
cv_results <- mandala_gp_cv(
GRM = GRM,
data = s2_data,
gen_col = "geno",
fixed = yld ~ env,
random = ~ GM(geno, Gmat),
R_formula = ~ vcov(RMAT),
R_matrix = RMAT,
k_folds = 5,
n_reps = 3
)Summary
TipKey Takeaways
- GRM integration at Stage 2 enables genomic prediction on BLUEs
- Use
GM(geno, Gmat)to specify genomic relationship structure - Combine with
vcov(RMAT)to properly weight BLUE uncertainty mandala_grm_prep()handles GRM alignment and scalingmandala_gp()provides a convenient wrapper for the full workflow- Genomic predictions are most valuable for untested genotypes and low-heritability traits
Next Steps
- Explore cross-validation with
mandala_gp_cv()to assess prediction accuracy - Consider factor-analytic models (
FA()) for complex G×E patterns - Review the Quick Reference for function details
Session Information
sessionInfo()R version 4.5.2 (2025-10-31)
Platform: aarch64-apple-darwin20
Running under: macOS Tahoe 26.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
locale:
[1] C.UTF-8/C.UTF-8/C.UTF-8/C/C.UTF-8/C.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Matrix_1.7-4 ggplot2_4.0.0 dplyr_1.1.4 mandala_1.1.0
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 cli_3.6.5 knitr_1.50 rlang_1.1.6
[5] xfun_0.54 generics_0.1.4 S7_0.2.0 splines2_0.5.4
[9] jsonlite_2.0.0 labeling_0.4.3 glue_1.8.0 htmltools_0.5.8.1
[13] scales_1.4.0 rmarkdown_2.30 grid_4.5.2 evaluate_1.0.5
[17] tibble_3.3.0 fastmap_1.2.0 yaml_2.3.10 lifecycle_1.0.4
[21] compiler_4.5.2 codetools_0.2-20 RColorBrewer_1.1-3 htmlwidgets_1.6.4
[25] Rcpp_1.1.0 pkgconfig_2.0.3 farver_2.1.2 lattice_0.22-7
[29] digest_0.6.37 R6_2.6.1 utf8_1.2.6 tidyselect_1.2.1
[33] pillar_1.11.1 magrittr_2.0.4 withr_3.0.2 gtable_0.3.6
[37] tools_4.5.2